


Βιοπληροφορική - Δεδομένα, Στατιστική και ο Κώδικας της Ζωής
ΑΦΙΕΡΩΜΑ - Η Νέα Εποχή στην Ιατρική

του ΠΑΝΑΓΙΩΤΗ ΠΑΠΑΣΤΑΜΟΥΛΗ,
Επίκουρου Καθηγητή Στατιστικής Μεθοδολογίας του Τμήματος Στατιστικής του ΟΠΑ
Η σύγχρονη βιολογία παράγει μεγάλο όγκο δεδομένων, ο οποίος είναι συγκρίσιμος με τις τάξεις μεγέθους που απαντώνται στη Σωματιδιακή Φυσική ή την Αστρονομία. Σε αυτό έχει συμβάλει κατά κύριο λόγο η Γενετική, δηλαδή το πεδίο της Βιολογίας που ασχολείται με τη μελέτη των γονιδίων, κληρονομικότητας και βιοποικιλότητας. Προβλήματα αυτού του είδους συνήθως αποτελούνται από δεκάδες χιλιάδες συνιστώσες (γονίδια, πρωτεΐνες κλπ), οι οποίες μπορεί να αλληλεπιδρούν μεταξύ τους, κάτι που μπορεί να κατανοηθεί επαρκώς μόνο με τη συλλογή μεγάλου όγκου δεδομένων και την ανάπτυξη/εφαρμογή κατάλληλων μεθοδολογικών εργαλείων για την ανάλυσή τους.
Για να εξερευνηθούν αυτά τα μονοπάτια, είναι απαραίτητος ο συγκερασμός πολλών επί μέρους επιστημονικών πεδίων, όπως η Υπολογιστική Επιστήμη, η Στατιστική, τα Μαθηματικά και φυσικά η Βιολογία. Ο κοινός αυτός διεπιστημονικός τόπος είναι γνωστός με τον όρο Βιοπληροφορική, ο οποίος άρχισε να αναπτύσσεται ραγδαία μετά το τέλος της δεκαετίας του 1990. Χρονικά, αυτό συμπίπτει με την ολοκλήρωση του διεθνούς ερευνητικού προγράμματος Human Genome Project, που οδήγησε στην αποκωδικοποίηση του ανθρώπινου γονιδιώματος.
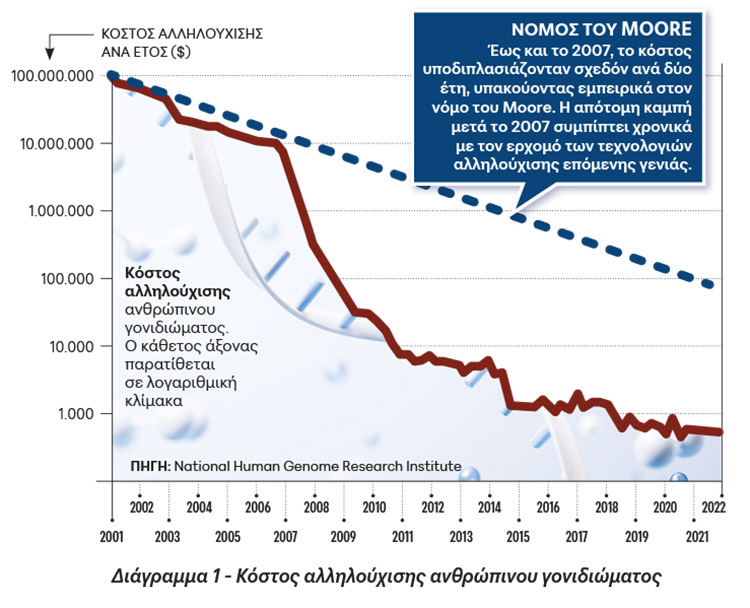
Ο αλματώδης, σχεδόν φρενήρης, ρυθμός εξελίξεων στον τομέα αυτόν, αποτυπώνεται χαρακτηριστικά στο Διάγραμμα 1 όπου παρατίθεται η διαχρονική εξέλιξη του κόστους αλληλούχισης του ανθρώπινου γονιδιώματος. Έως και το 2007, το κόστος υποδιπλασιάζονταν σχεδόν ανά δύο έτη, υπακούοντας εμπειρικά στον νόμο του Moore. Η απότομη καμπή μετά το 2007 συμπίπτει χρονικά με τον ερχομό των τεχνολογιών αλληλούχισης επόμενης γενιάς.
Η περιοχή της Βιοπληροφορικής ανέδειξε το πρόβλημα του «μεγάλου p και μικρού n» δηλαδή της ύπαρξης μικρού αριθμού παρατηρούμενων υποκειμένων (π.χ. ανθρώπων) και μεγάλου αριθμού χαρακτηριστικών τους. Aς θεωρήσουμε ότι επιθυμούμε να προβλέψουμε την παρουσία ή όχι μιας συγκεκριμένης ασθένειας με βάση ένα δείγμα n = 200 ατόμων, όπου σε κάθε ένα καταγράφεται η έκφραση p = 20.000 γονιδίων. Τα κλασικά προβλεπτικά μοντέλα αντιμετωπίζουν μία σειρά προβλημάτων σε αυτές τις περιπτώσεις, γιατί ο αριθμός των παραμέτρων που πρέπει να εκτιμηθούν είναι πολύ μεγαλύτερος από το μέγεθος του δείγματος. Οι Στατιστικοί τότε συνειδητοποίησαν ότι μπορούν να παρακάμψουν αυτά τα προβλήματα προτείνοντας εναλλακτικές τεχνικές που υποστηρίζουν απλούστερα μοντέλα που εμπλέκουν λιγότερα χαρακτηριστικά για την πρόβλεψη.
Η ποσοτικοποίηση της γονιδιακής έκφρασης – δηλαδή του βαθμού που ένα γονίδιο απαντάται σε ένα δείγμα – είναι ένα βασικό ζήτημα στην μοριακή βιολογία. Ένας από τους κύριους στόχους των επιστημόνων είναι να βρουν ποια γονίδια διαφοροποιούνται μεταξύ δύο ή και περισσότερων ομάδων (πχ υγειών έναντι ασθενών). Για να το πετύχουν, μετρούν τα επίπεδα ενός συγκεκριμένου μορίου που παράγεται από τα γονίδια, του αγγελιοφόρου RNA, χρησιμοποιώντας εξειδικευμένες τεχνικές, όπως οι μικροσυστοιχίες (microarrays) και η πιο σύγχρονη τεχνική RNA-Sequencing. Ένα τέτοιο πείραμα παράγει τεράστιο όγκο δεδομένων – από δεκάδες έως και εκατοντάδες εκατομμύρια μικρά κομμάτια γενετικού υλικού.
Ένα τυπικό πείραμα RNA-sequencing παράγει τεράστιο όγκο δεδομένων – από δεκάδες έως και εκατοντάδες εκατομμύρια μικρά κομμάτια γενετικού υλικού (μικρές ακολουθίες νουκλεοτιδικών βάσεων). Στη συνέχεια, αυτά πρέπει να τοποθετηθούν στη σωστή θέση μέσα στο DNA, κάτι που είναι εξαιρετικά περίπλοκο, αφού κάθε μικρό κομμάτι μπορεί να ταιριάζει σε πολλά διαφορετικά σημεία. Για να λυθεί αυτό το πρόβλημα, οι επιστήμονες χρησιμοποιούν προχωρημένα υπολογιστικά εργαλεία και στατιστικές μεθόδους.
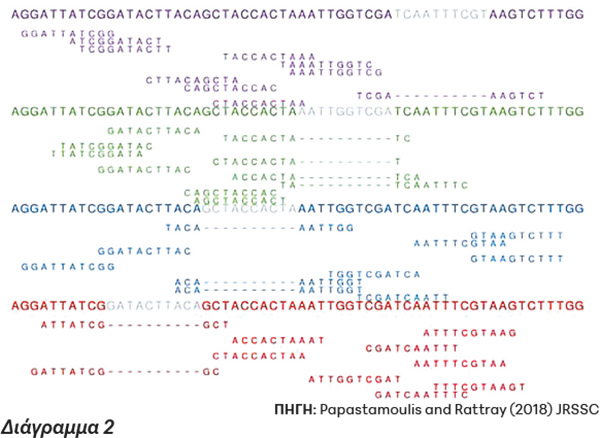
Απλοποιημένο παράδειγμα RNA-sequencing data:
μικρές ακολουθίες βάσεων που πρέπει να στοιχιστούν στα αντίστοιχα τέσσερα γονίδια (μεγαλύτερες ακολουθίες βάσεων). Το χρώμα αντιστοιχεί στην προέλευση κάθε μικρής ακολουθίας αλλά στην πραγματικότητα δεν το γνωρίζουμε αυτό και πρέπει να το εκτιμήσουμε. Πηγή: Papastamoulis and Rattray (2018) JRSSC
Στο Διάγραμμα 2 παρατίθεται μία απλοποιημένη μορφή ενός τέτοιου παραδείγματος. Σκοπός της ανάλυσης είναι να εκτιμηθεί ο αριθμός των δεδομένων που έχει παράξει κάθε ένα από τα (τέσσερα) γονίδια χωρίς βέβαια να γνωρίζουμε την πραγματική τους προέλευση (στο Διάγραμμα 2 αυτό φαίνεται με το αντίστοιχο χρώμα). Η δυσκολία έγκειται στο γεγονός ότι οι περισσότερες μικρές ακολουθίες στοιχίζονται το ίδιο καλά σε παραπάνω από ένα γονίδια, οπότε ο αριθμός των ακολουθιών που έχουν προέλθει από καθένα από τα 4 γονίδια είναι αβέβαιος και πρέπει να εκτιμηθεί με κατάλληλα στατιστικά μοντέλα.
Η κοινότητα της βιοπληροφορικής βασίζεται σε μεγάλο βαθμό σε λογισμικό ανοιχτού κώδικα και λειτουργικά συστήματα όπως το Linux. Για την ανάπτυξη αλγορίθμων που απαιτούν βελτιστοποιημένη διαχείριση μνήμης και γρήγορη εκτέλεση προτιμάται συχνά η C++, ενώ για ανάλυση δεδομένων και οπτικοποίηση κυριαρχούν οι γλώσσες R και Python.
Εν κατακλείδι, η ραγδαία ανάπτυξη της βιοπληροφορικής δεν είναι απλώς τεχνολογική εξέλιξη, αλλά μια επανάσταση στον τρόπο που κατανοούμε τη ζωή. Η συνδυαστική δύναμη της στατιστικής, της υπολογιστικής επιστήμης και της βιολογίας επιτρέπει την εξαγωγή πολύτιμων γνώσεων από τεράστια σύνολα δεδομένων, φέρνοντας την επιστήμη πιο κοντά σε εξατομικευμένες θεραπείες και νέες βιοϊατρικές ανακαλύψεις. Παρόλα αυτά, οι προκλήσεις παραμένουν: η διαχείριση και η ανάλυση μεγάλων δεδομένων απαιτούν συνεχή εξέλιξη των μεθόδων και των εργαλείων. Στο μέλλον, η πρόοδος θα εξαρτηθεί από τη διεπιστημονική συνεργασία και την ανάπτυξη ακριβέστερων μοντέλων, διαμορφώνοντας έναν κόσμο όπου η πληροφορία γίνεται θεραπεία και η στατιστική αποκαλύπτει τα μυστικά της ζωής.